许多大佬的软件想要构建一个大而美的生态,从 monocle2 开始就能做单细胞的质控、降维、分群、注释这一系列的分析,但不幸的是我们只知道 monocle 系列还是主要做拟时序分析,一方面是因为 Seurat 有先发优势,出名要趁早,生态太过强大,另一方面 monocle2 和monocle3 软件有自身的问题,很难安装或者有很多 bug,远没有 Seurat 稳定。

什么是Monocle3?
不过怎么样Monocle3 还是最常用的单细胞轨迹分析工具之一,它能够通过算法学习细胞在动态生物学过程中基因表达变化的序列,从而构建出细胞状态转变的轨迹。与传统实验方法不同,Monocle3无需纯化处于中间状态的细胞,就能让我们清晰地看到细胞从一种功能“状态”向另一种状态的过渡。
当细胞过程存在多种结果时,Monocle3会构建出“分支”轨迹,这些分支对应着细胞的“决策”过程。它还提供了强大的工具来识别受这些决策影响以及参与决策的基因,帮助我们深入理解细胞命运决定的分子机制。
Monocle3核心思想
无需通过实验将细胞提纯为离散状态,而是借助算法学习细胞在动态生物学过程中必然经历的基因表达变化序列,进而掌握基因表达变化的整体 “轨迹”,并确定每个细胞在该轨迹中的准确位置。
核心工作流程
Monocle3的轨迹重建工作流程与聚类分析流程相似,但增加了几个关键步骤。下面我们详细介绍其核心流程:
安装
注意 linux、mac 安装都需要编译,那么就需要 gcc、gfortran等编译软件,确保已经安装
sudo apt install build-essential pkg-config libgdal-dev -y
BiocManager::install(c('BiocGenerics', 'DelayedArray', 'DelayedMatrixStats','limma', 'lme4', 'S4Vectors', 'SingleCellExperiment','SummarizedExperiment', 'batchelor', 'HDF5Array','ggrastr'))
BiocManager::install("bnprks/BPCells/r")
BiocManager::install('cole-trapnell-lab/monocle3')
library(monocle3)
1. 数据准备与初始化
首先,需要将数据加载到Monocle3的cell_data_set(CDS)对象中,这是Monocle3处理数据的基本单位。CDS对象包含三个关键部分:表达矩阵、细胞元数据和基因注释。
官网示例
library(monocle3)
# 加载数据
expression_matrix <- readRDS(url("https://depts.washington.edu:/trapnell-lab/software/monocle3/celegans/data/packer_embryo_expression.rds"))
cell_metadata <- readRDS(url("https://depts.washington.edu:/trapnell-lab/software/monocle3/celegans/data/packer_embryo_colData.rds"))
gene_annotation <- readRDS(url("https://depts.washington.edu:/trapnell-lab/software/monocle3/celegans/data/packer_embryo_rowData.rds"))# 创建CDS对象
cds <- new_cell_data_set(expression_matrix,cell_metadata = cell_metadata,gene_metadata = gene_annotation)
但是更多时候我们是从已经处理好的 Seurat 对象构建CDS对象
# srt是之前注释好的Seurat 对象
cds <- monocle3::new_cell_data_set(expression_data = SeuratObject::GetAssayData(srt, assay = "RNA", layer = "counts"),cell_metadata = srt@meta.data,gene_metadata = data.frame(gene_short_name = rownames(srt), row.names = rownames(srt))
)
2. 数据预处理
预处理步骤与聚类分析完全相同,包括数据标准化和批次效应校正等。在批次校正中,可以使用align_cds()函数,并通过alignment_group指定批次分组
# 数据预处理
cds <- preprocess_cds(cds, num_dim = 50)
# 批次校正
cds <- align_cds(cds, alignment_group = "batch")
3. 降维与可视化
接下来进行数据降维,对于轨迹分析,强烈建议使用UMAP方法(默认方法)。降维后可以使用plot_cells()函数可视化结果,通过不同的颜色编码展示细胞类型等信息。
# 降维
cds <- reduce_dimension(cds)用 Seurat 对象中的 UMAP 替换 CDS 中的
cds.embed <- cds@int_colData$reducedDims$UMAP
int.embed <- Embeddings(srt, reduction = "umap")
int.embed <- int.embed[rownames(cds.embed), ]
cds@int_colData$reducedDims$UMAP <- int.embed
可视化 UMAP 图
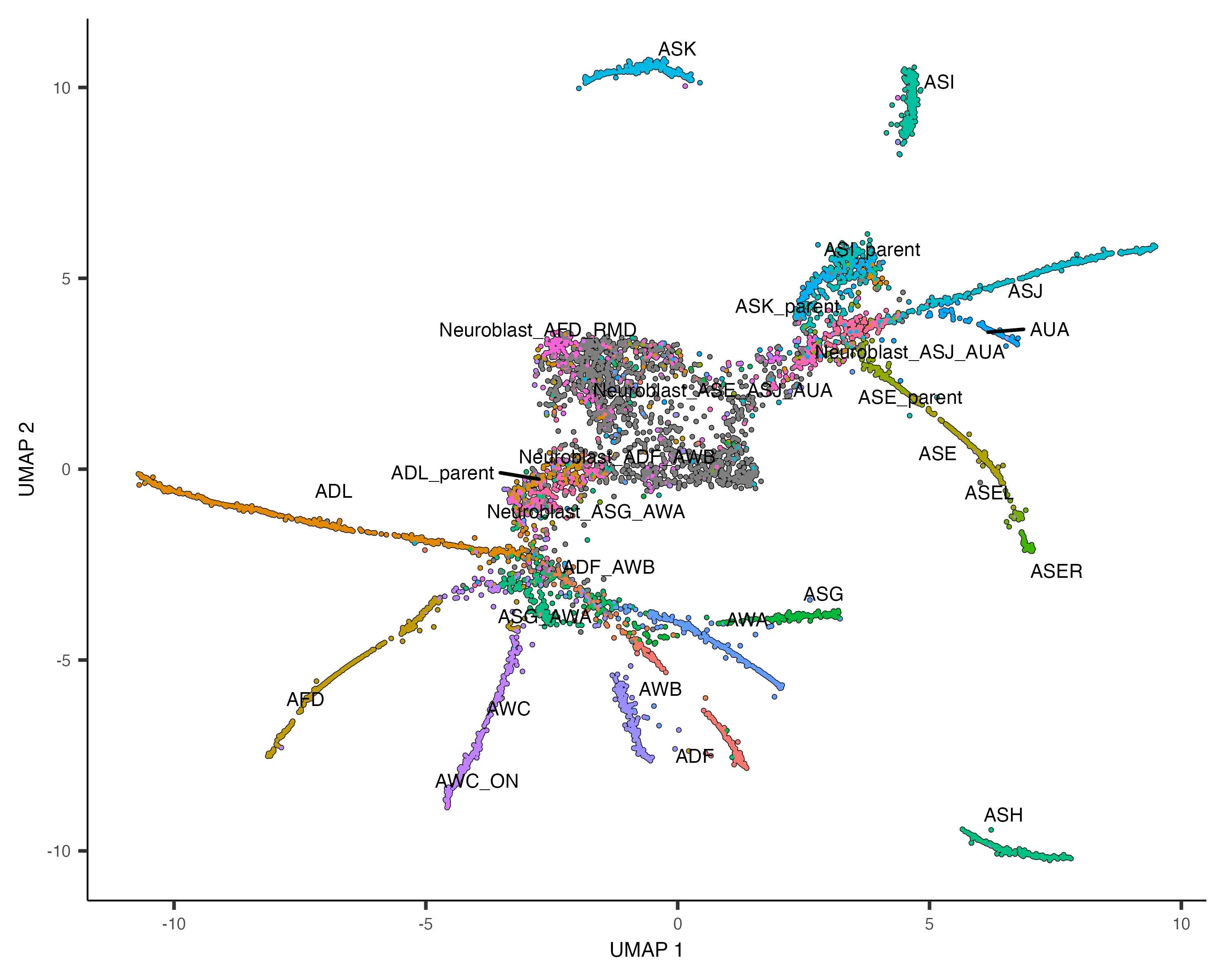
plot_cells(cds, label_groups_by_cluster = FALSE, color_cells_by = "cell.type")
通过这一步骤,我们可以初步观察到数据中存在的轨迹结构,以及不同细胞类型在轨迹上的分布。
4. 细胞聚类
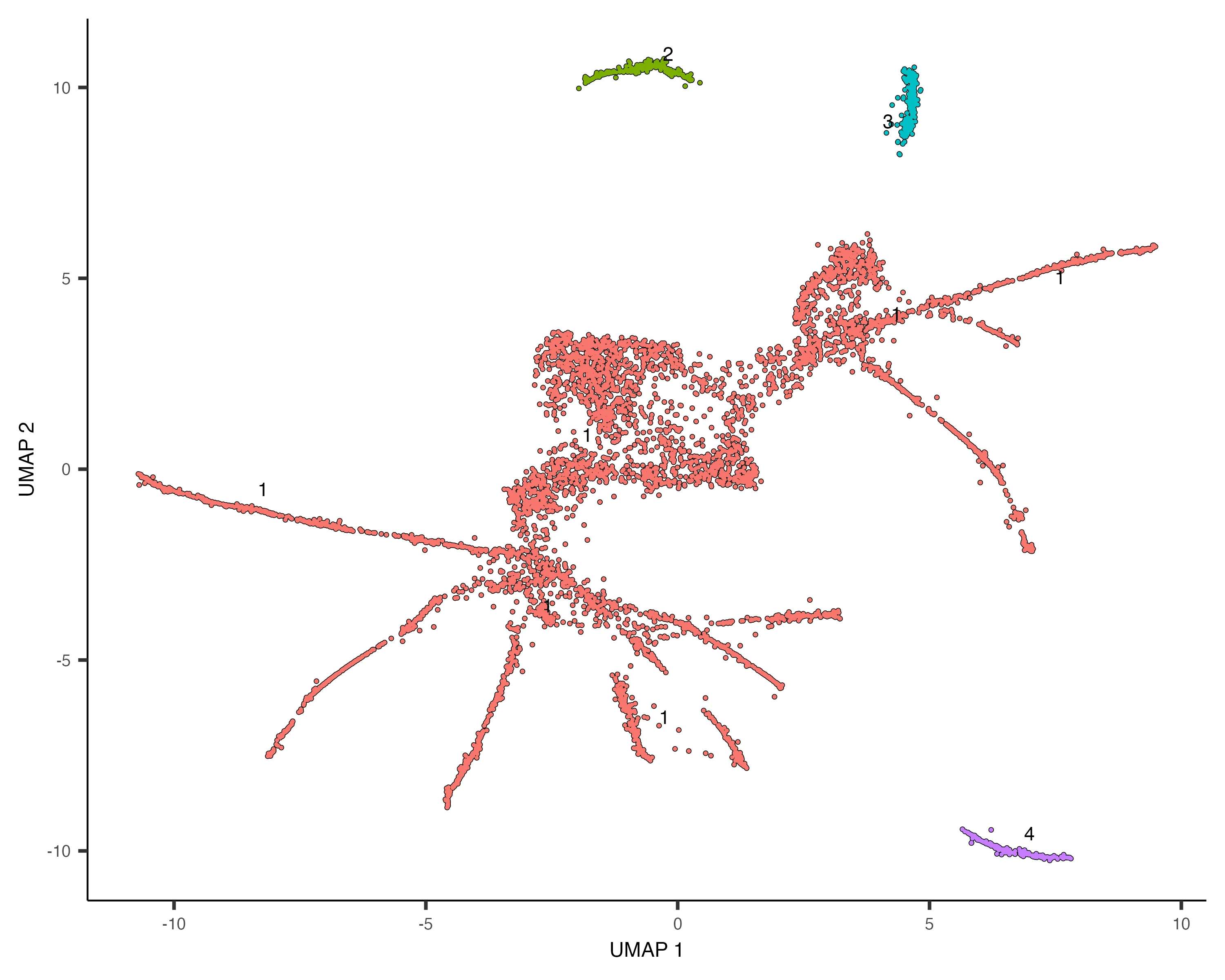
与聚类分析类似,使用cluster_cells()函数对细胞进行聚类。在轨迹分析中,每个细胞不仅被分配到一个聚类,还会被分配到一个分区(partition),每个分区最终会形成一个独立的轨迹。
不同的分区可以有不同的分化起始点,这也符合现实情况,因为很可能这些细胞是多起源的。
# 细胞聚类
cds <- cluster_cells(cds)
# 按分区可视化
plot_cells(cds, color_cells_by = "partition")
5. 学习轨迹图
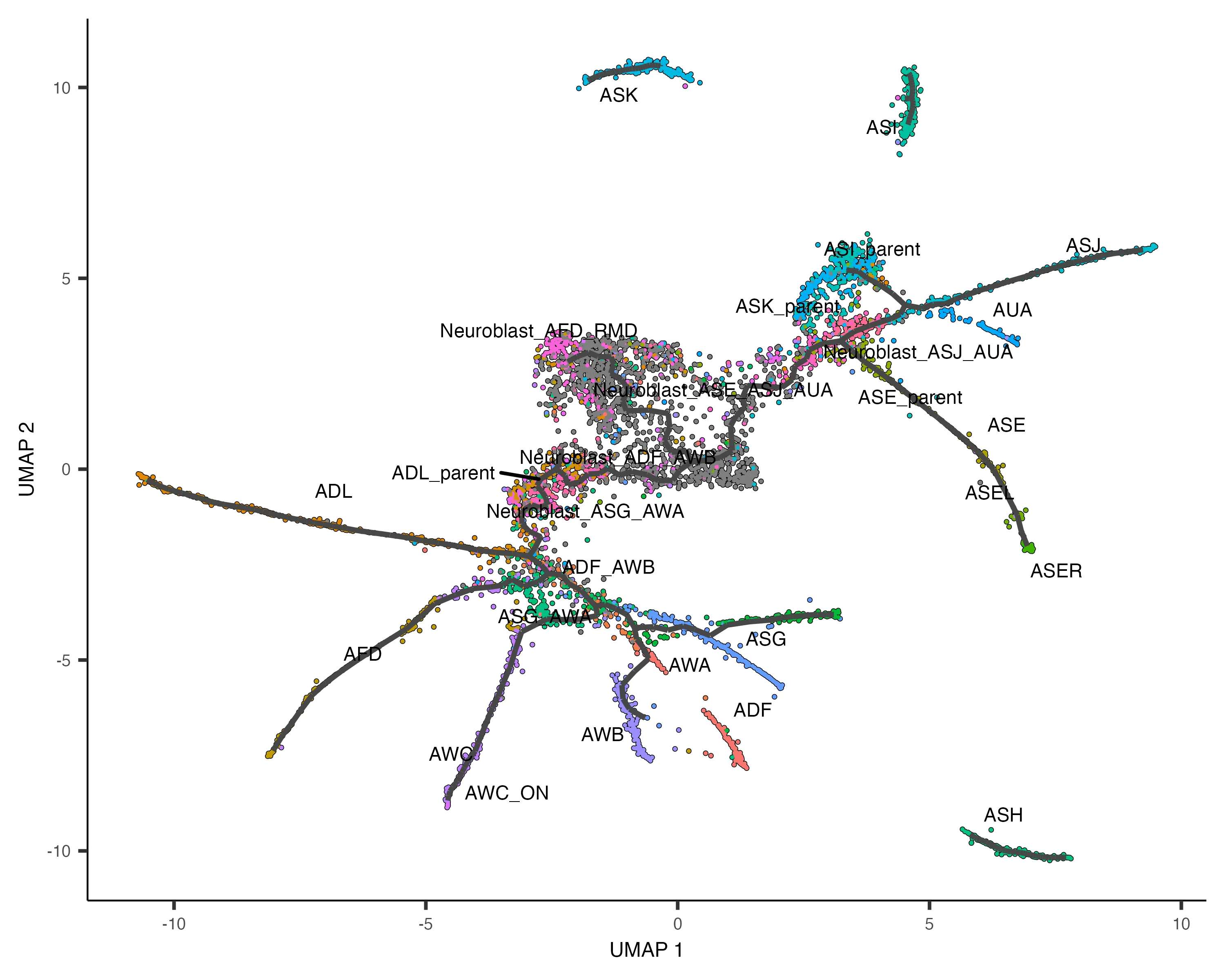
使用learn_graph()函数在每个分区内拟合主图(principal graph),该图将用于后续的分支分析和差异表达分析等下游步骤。
# 学习轨迹图
cds <- learn_graph(cds)
# 可视化轨迹图
plot_cells(cds, color_cells_by = "cell.type", label_groups_by_cluster = FALSE, label_leaves = FALSE, label_branch_points = FALSE)
轨迹图的构建是Monocle3轨迹分析的核心步骤之一,它能够捕捉细胞状态之间的连续过渡关系。
6. 以伪时间(Pseudotime)排序细胞
伪时间是衡量单个细胞在细胞分化等过程中进展程度的指标。通过order_cells()函数,我们可以根据细胞在轨迹上的进展对其进行排序。
- 什么是伪时间? 伪时间是一个抽象的进展单位,它表示细胞沿着学习到的轨迹从起始状态到结束状态所经历的距离。在许多生物学过程中,细胞的发育并不同步,伪时间很好地解决了这种不同步发育带来的分析难题。
# 按伪时间排序细胞
cds <- order_cells(cds)
# 按伪时间可视化
plot_cells(cds, color_cells_by = "pseudotime", ...)
在排序过程中,需要指定轨迹图的根节点(root nodes),可以通过图形界面手动选择,也可以通过编程方式指定。例如,可根据最早时间点的细胞分布来自动确定根节点:
下边这个函数可以帮助我们选择发育起点,比如我们可以修改为从某一类细胞发育作为 root,只需要修改embryo.time.bin 为 cell.type 和time_bin 为我们想要设定的细胞类型
# 编程方式指定根节点
get_earliest_principal_node <- function(cds, time_bin="130-170"){cell_ids <- which(colData(cds)[, "embryo.time.bin"] == time_bin)closest_vertex <-cds@principal_graph_aux[["UMAP"]]$pr_graph_cell_proj_closest_vertexclosest_vertex <- as.matrix(closest_vertex[colnames(cds), ])root_pr_nodes <-igraph::V(principal_graph(cds)[["UMAP"]])$name[as.numeric(names(which.max(table(closest_vertex[cell_ids,]))))]root_pr_nodes
}
cds <- order_cells(cds, root_pr_nodes = get_earliest_principal_node(cds))
值得注意的是,未从根节点可达的细胞会被分配无限伪时间(显示为灰色),因此通常应为每个分区至少选择一个根节点。
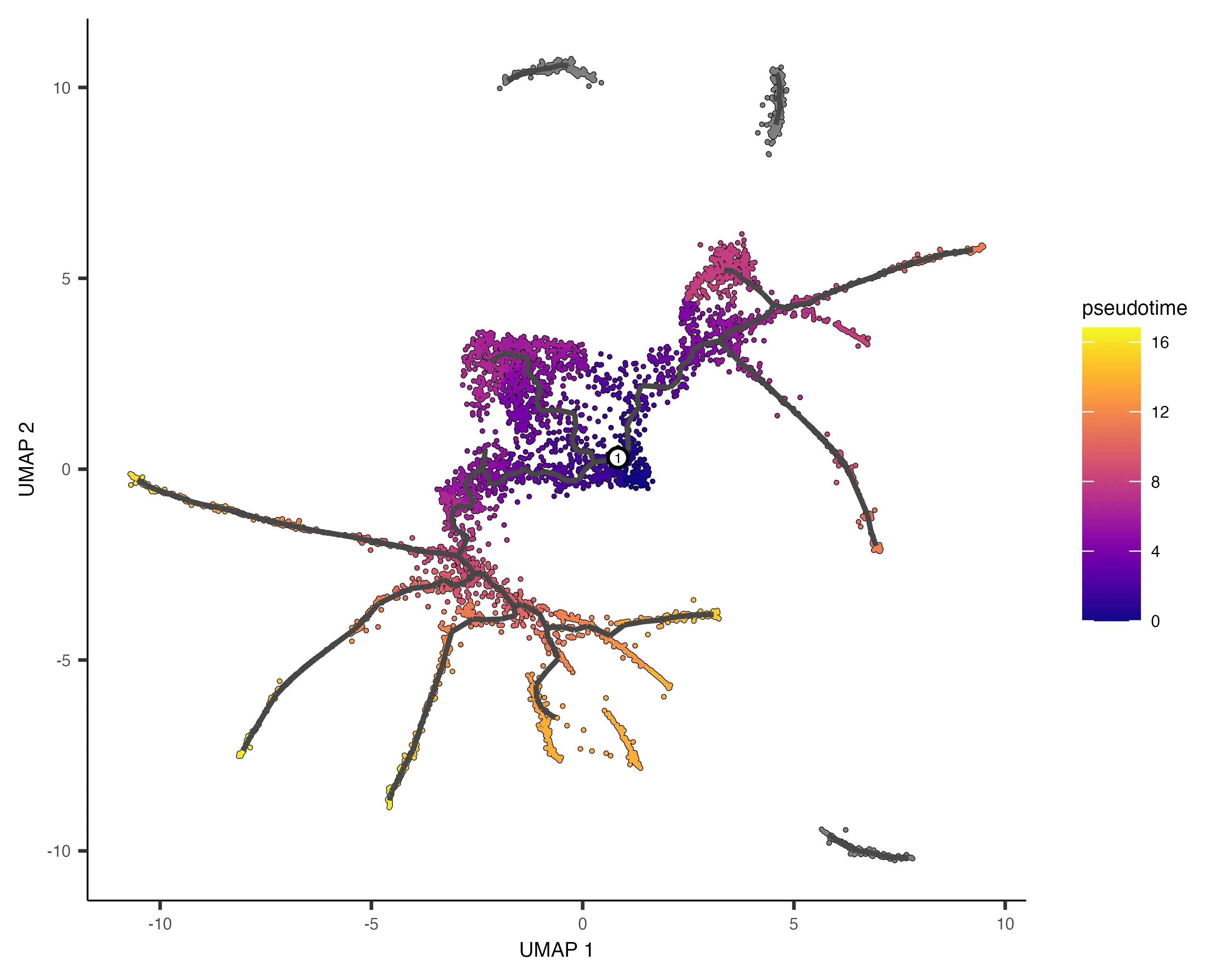
7. 时序相关差异表达分析
Monocle3提供了强大的差异表达分析工具,可用于寻找随着伪时间变化或在不同分支中表达发生变化的基因。例如:
可以选择前 10 个基因展示,官网教程是自己选择的与研究相关的基因
genes <- monocle3::graph_test(cds, neighbor_graph = "principal_graph", reduction_method = "UMAP", cores = 32)
top10 <- genes %>%top_n(n = 10, morans_I) %>%pull(gene_short_name) %>%as.character()
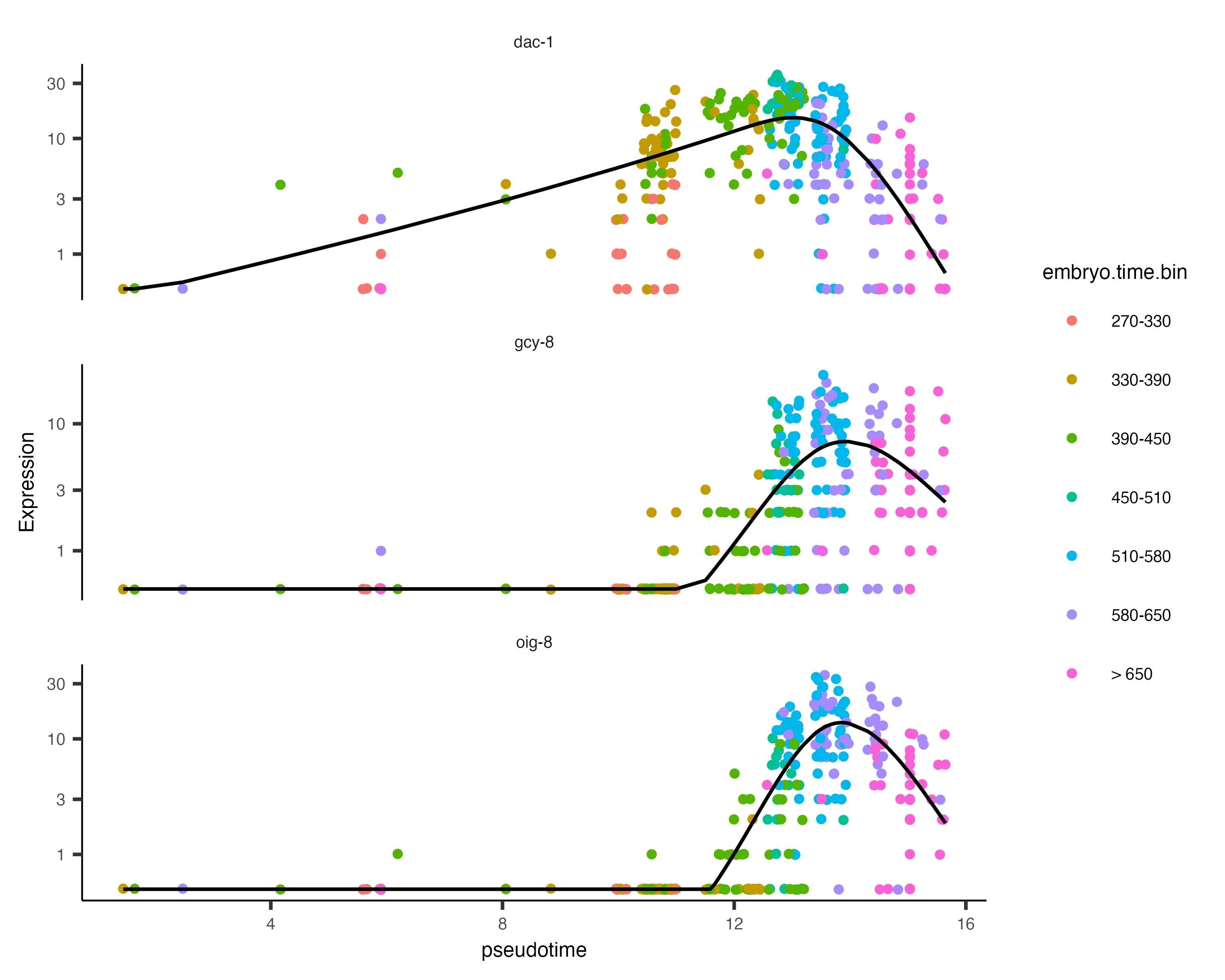
展示AFD_genes 基因随时间变化图
AFD_genes <- c("gcy-8", "dac-1", "oig-8")
AFD_lineage_cds <- cds[rowData(cds)$gene_short_name %in% AFD_genes,colData(cds)$cell.type %in% c("AFD")]
AFD_lineage_cds <- order_cells(AFD_lineage_cds)plot_genes_in_pseudotime(AFD_lineage_cds,color_cells_by="embryo.time.bin",min_expr=0.5)
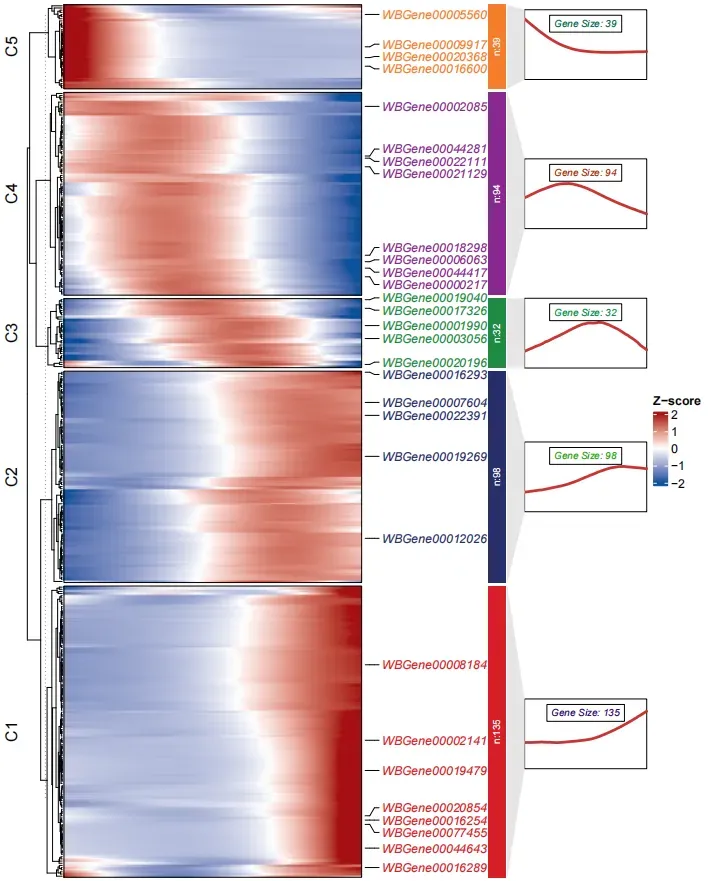
8. 拟时序基因热图
这里演示下选择 50 个基因画热图用 junjun 老师的 R 包ClusterGVis绘制,虽然 monocle3 有自己的分模块热图,感觉没啥用也不好看,不如这个更醒目一些。
top50 <- genes %>%top_n(n = 10, morans_I) %>%pull(gene_short_name) %>%as.character()
绘制热图,cluster.num 分 5 个 cluster 需要自己设置,markGenes 随机选择 30 个展示,可以自己设置标注哪些基因
library(ClusterGVis)# kmeans
ck <- clusterData(mat,cluster.method = "kmeans",cluster.num = 5)# add line annotation
pdf('monocle3.pdf',height = 10,width = 8,onefile = F)
visCluster(object = ck,plot.type = "both",add.sampleanno = F,markGenes = sample(rownames(mat),30,replace = F))
dev.off()
Reference
https://cole-trapnell-lab.github.io/monocle3/docs/differential/
https://mp.weixin.qq.com/s/7sYTrHlhnHj5490RyrdBug


介绍)











![[工具类] 网络请求HttpUtils](http://pic.xiahunao.cn/[工具类] 网络请求HttpUtils)
)
变换矩阵中的齐次坐标推导与几何理解)


